Generating
multiple
sequence alignments and trees with Clustal, Phylip and PAUP
You are being
provided with three data sets for
analysis. Two of the data sets are
protein and one is nucleotide. This assignment has 5 parts.
1.
Making
multiple sequence alignments (MSAs) for your data sets (or you
can use alignments created with Urmilla).
2.
Studying
your alignments to see what you can learn from them
3.
Use
MSAs for phylogenetic analysis with Phylip
- Distance
- Parsimony
4.
Use
MSAs for bootstrap analysis
5.
Interpret
results.
Before you
begin, prepare your work space:
Make a new
directory for this assignment "mkdir phylo" Change into this new directory and make three additional directories. If you are
using alignments created with Urmilla, make directories for these
files. One for each MSA. Label them with the data type, e.g. nucleotide
or protein
cd phylo
mkdir cs-prot
mkdir actin-prot
mkdir actin-nuc
Now, "cd"
into each of these directories
and copy over the original text
files used to make the MSA with clustalX and the resulting phylip
formatted
alignment file from wherever you put the files from Urmilla's ecercise.
Or, download the files provided to you on the web. You will find thm in
the data directory. Be sure to put the files in the correct
directories that you created for them.
cd cs-prot
download
cs.txt
download
cs.phy
cd ../actin-prot
download
actinpep.txt NOTE: there is no
alignment you will need to create it
cd ../actin-nuc
download
actinnuc.txt NOTE: there is no
alignment you will need to creat it
ClustalX takes a text file of fasta formatted
sequences and
creates an interleaved multiple sequence alignment out of them. You
have
control over the output sequence formats. By default the program will
create
".aln" and ".dnd" formats. You will need to add the
".phy" formats. Do this within
the correct directories. This should take only 4-5 minutes each.
Launch
clustalX by typing "clustalx" on the command line .
- When
the program opens, you can input the sequences to be aligned by
clicking on "file" in the menu bar and selecting load sequences. Load your sequence.txt file.
- Now,
click on "Alignment" in the menu bar and select the last opion "Output
format
options" and select "clustal" and "phylip".
- Now click on "alignments" in the menu bar
and select "do complete alignment". The
program will show you where it is planning to locate the
output
files, make sure the paths are correct and then click on "align"
2)
Study your alignments. What do you see? Please answer the following questions
about each of your alignments (except question ÒeÓ which
only applied to one
data set).
The actin data sets contain the same sequences,
as either DNA or protein. Yeast is
your outgroup. The drosophila sequences represent the protostome
organisms and
the all of the orther organisms are deuterostomes, with tunicates
(halocynthia)
and Echinoderms (strongylocentrotus purpuratus) as representatives of
the early
branches. You are now ready to begin.
Please perform all analyses related to a particular data set
within the
directory for that data set.
1.
Are
there lots of gaps?
2.
Is
this a highly conserved protein or DNA sequence?
3.
Are
the sequences used to make the
alignment of roughly equal length? Explain.
4.
Do
you see any significant shared insertions or deletions among
phylogenetically
related sequences
5.
For
the actin-nuc data set only, does the nucleotide alignment correspond
to the aa
alignment?
3 & 4) Phylogenetic
analysis. We will explore the use of
Phylip and two different phylogenetic
reconstruction methods (Distance and Parsimony). Phylip is free and
available for
many platforms, but it is a bit tedious to use.
Documentation
for each of the phylip programs can be found in the
documentation folder located with the Phylip programs. Ask the
course assistants to find out where these are located. The
documentation file names for the phylip
programs are "seqboot.doc" or "neighbor.doc" etc...
Phylogenetic analysis with
Phylip (Phylip is a
collection
of approx 30 programs). We will
only use 7 of them for this exercise.
Protdist -
for generating a distance matrix with
protein sequence
Protpars
- for generating parsimony trees with
protein sequence
Dnadist
- for generating a distance matrix with
DNA sequences
Dnapars
- for generating parsimony trees with
DNA sequences
Neighbor -
for generating either a
neighbor-joining or UPGMA tree from a specified distance matrix (DNA or
protein)
Seqboot
- for generating bootstrapped data sets
(any type of sequence)
Consense
- for generating consensus trees
LOOK AT
THE DESCRIPTIONS OF EACH PROGRAM LOCATED BELOW FOR MORE
INFORMATION
As
demonstrated in the workshop, you will need
to pass your multiple sequence alignment through a series of programs.
Each is
step is quick. The programs are used in the following order:
Protdist
- neighbor To
construct protein distance tree
Dnadist
- neighbor To
construct DNA distance tree
Dnapars
To
construct DNA parsimony tree(s)
Protpars
To
construct Protein parsimony tree(s)
After
completing the above and saving the data
to files with new names, you can perform a bootstrap analysis of any of
the
above 4 scenarios, by first using seqboot (use 10 replicates for this
exercise
so that things will go quickly, but in real life choose at least 100
replicates), and in the end, use
consense on your treefile. e.g. seqboot - protdist -
neighbor - consense.
Don''t forget to select the multiple option on each program in
a bootstrap analysis, it must know how many data sets to expect.
The input for
the first (or in the case of
parsimony the only) program in each series is always your alignment in
phylip
format (file.phy renamed as infile).
ALL programs read "infile" whatever that may be (either your
alignment
or the output from an earlier step) except for consense, it reads
treefile.
All of your
trees (treefile) can be viewed and
edited in "treetool" and printed from there ( NOTE: Treetool may
not be installed, you may need to get and install the program.
How would you do that?.
Remember!
Each phylip program only
accepts infile and produces only outfile and/or treefile. Keep copies
of each
of these files with different names before moving on to the next step
or you
will have to begin again!. For example.
My multiple sequence alignment was rpl2.phy
As an example I did the following:
cp cs.phy infile (makes
a copy of your alignment to
use for phylip)
protdist
(Makes
protein distance calculations from infile)
cp outfile
protdist.out
(save
a copy of output)
cp outfile infile
(copy output
into infile
for next program)
neighbor
(makes
tree from distances)
cp outfile
neighbor.out (save a copy
of output from neighbor) neighbor will also create a treefile
In the end,
all you will need to keep is the
file cs.phy (the alignment file) protdist.out (the distance matrix)
neighbor.out (your branch lengths and groups) and treefile (your tree).
Look at the
outfile generated at each of these
steps. It will soon become very clear to you what is happening at each
step.
Also, play with the parameters offered in each program it only takes
two
minutes to see the results. What happens if you use a
matrix other than PAM for protdist?
You can
download the programs or source code or
documentation from http://evolution.genetics.washington.edu/phylip.html
if you are interested.
Successful completion of
this exercise requires that you
generate and think about the
following;
Three
multiple sequence alignments: one for CS and two for Actin (one DNA and
one
Protein)
1.
The
answers to the questions about the alignments listed above (a-d or a-e)
For
the cs-prot data set, perform the following analyses:
1.
Distance
(neighbor-joining) analysis with Phylip
2.
Parsimony
analysis with Phylip
For
the actin-prot data set, perform the following analyses:
1.
Distance
(neighbor-joining) analysis with Phylip
2.
Bootstrap
of the above analysis with Phylip
For
the actin-nuc data set, perform the following analyses:
1.
Distance
(neighbor-joining) analysis with Phylip
2.
Parsimony
analysis with Phylip
For EACH
tree, note the method(s) and
relevant distance matrices or TS/TV ratios used to generate them
(perhaps in a
README file saved in the same directory as the trees)
Think
about the following:
1.
Did
the different phylogenetic methods that you tried (distance and
parsimony)
yield consistent results when used on the same MSA?
Did you get exactly the same tree each time?
2.
If
you look at the actin protein trees, what do they tell you about the
evolution
of actin?
3.
If
you compare your DNA and Protein trees for actin, do they give the same
trees? If not why you think the
results may have differed.
4.
Could
you tell, just by looking at your multiple sequence alignments roughly
how the
trees would turn out? What did you
see that gave you your hunch or idea about the relationships? Was your hunch correct?
5.
Turning
from molecular systematics to molecular evolution, what interesting
things
happened in the evolution of the circumsporozoite protein?
EXTRA INFORMATION
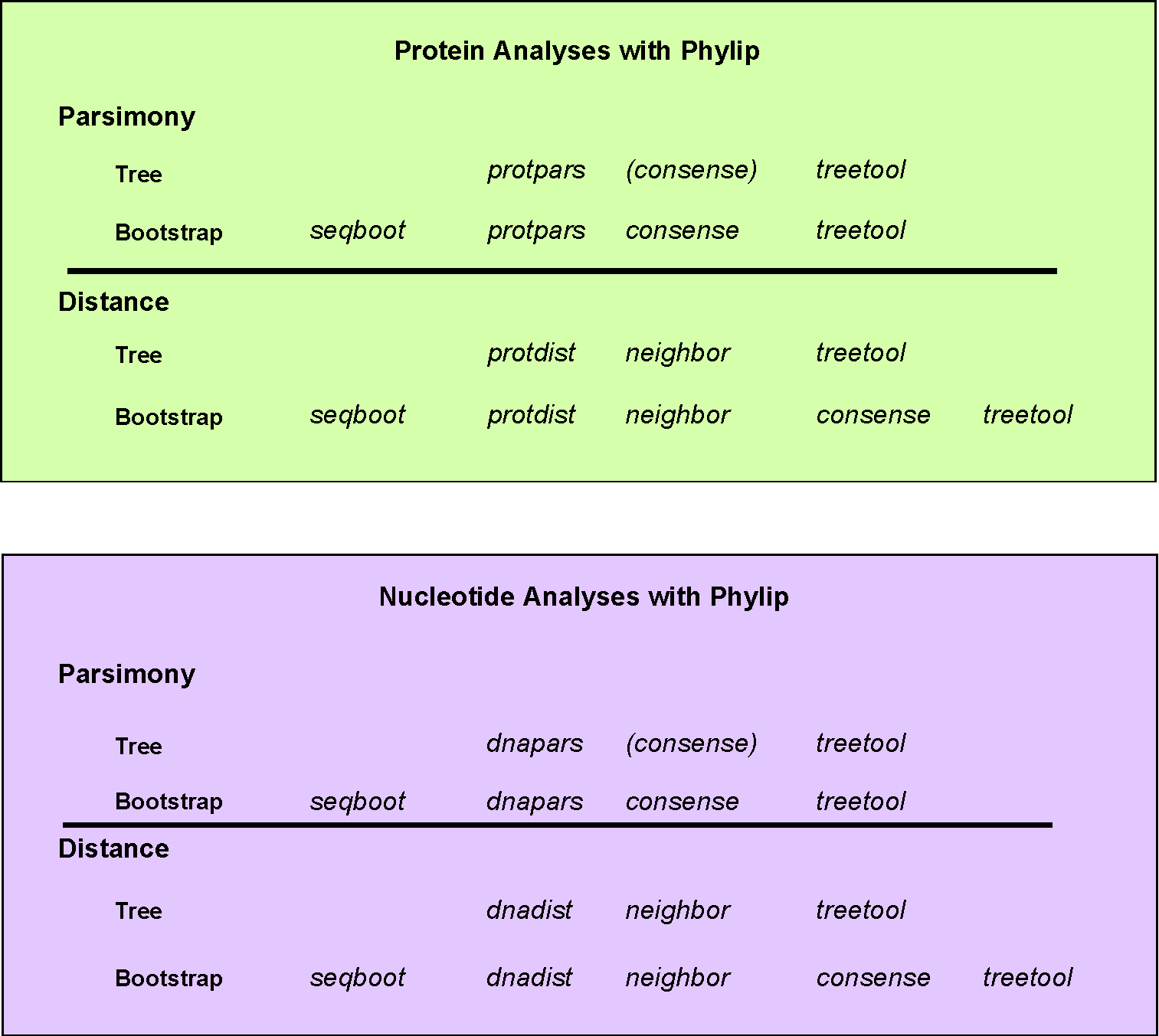
PHYLIP is a collection of programs, each of which performs a special task. You must call a new program for each task you want to perform. The programs relevant to this exercise are: seqboot, dnadist, protdist, neighbor, dnapars, protpars, consense. Each of these programs only accepts a file called infile by default (you can override) and each produces a file called outfile and/or treefile. WARNING: Each of these programs will overwrite any existing outfiles or treefiles present in the same directory!
Seqboot makes bootstrapped datasets (i.e. alignments) that can be used in ANY analysis. You must specify the number of datasets to make. You only need this program if you are performing a bootstrap analysis. It will create a single outfile.
Dnadist and Protdist are used to make distance matrices from DNA and protein data respectively. You must choose the weighting scheme and whether or not the data are interleaved. If your infile happens to be the output of seqboot, you need to select the option indicating that multiple datasets are in use.
Neighbor is one of many programs that will make a tree from the data matrices created by dnadist and protdist. This program will create both an outfile and a treefile. If you are running neighbor on a distance matrix file that originated from a seqboot set, i.e. a bootstrap, you need to select the option indicating that multiple datasets are being used.
Consense is only used on the treefile generated at the end of a bootstrap analysis. It will compute a consensus tree from your bootstrap trees. The input for consense is the treefile output generated by either dnadist, protdist, dnapars, or protpars.
Dnapars and Protpars perform parsimony analyses on dna and protein data respectively. These programs use the .phy multiple sequence alignment file as their infile.. If you need a parsimony bootstrap you must use seqboot first. Dnapars and protpars will generate an outfile and a treefile. If there are multiple trees or if you are doing a bootstrap analysis, you will need to use consense to get a consensus tree.
Once you have a treefile with a single tree in it, you can view that tree and edit it in treetool. You can launch the program by typing “treetool”. You can print you trees from treetool in many formats. You can also change the outgroup at this stage if you didn’t specify it correctly earlier and you can re-label branches with taxonomic names.
Examples:
If you want to perform a distance analysis on a DNA data set you would do the following: dnadist then neighbor then treetool
If you want to perform a bootstrapped distance analysis on a protein data set you would do the following: seqboot then protdist then neighbor then consense then treetool.
A bootstrapped parsimony analysis on a protein data set would look like: seqboot then protpars then consense then treetool.